The high cost of the human genome project, with 20 year time-line for completion in the year 2000, caused many livestock researchers to despair that reference genomes would not be available for their species in their lifetime. Due to incredible advancements in DNA sequencing technology, we are now able to generate reference-quality genome assemblies at a small fraction of the cost and with far less manual editing than in the past. While the process still isn’t completely trivial, we are now far better equipped to generate quality genome assemblies at scale due to the following technological improvements:
Longer reads == better genomes
The first method for sequencing bases in DNA molecules was developed by Fred Sanger in 1977. His method of sequencing was the predominant method from that year until the year 2005, when the first of the second-generation sequencing methods (pyro-sequencing) was developed and released. While this was a major improvement in the amount of bases that a single experiment could yield at a time, the method did not appreciably increase the length of individual DNA sequencing bases. In fact, it wasn’t until 2010 that the DNA sequencing read length “barrier” was broken by the use of new techniques to study single DNA molecules during replication. This innovation was followed up in 2015 by the release of new methods to sequence DNA using nanopores, which resulted in the longest DNA sequence reads ever generated by a single molecule. The following image shows a comparative range of DNA sequencing read technologies ordered by their chronological release:
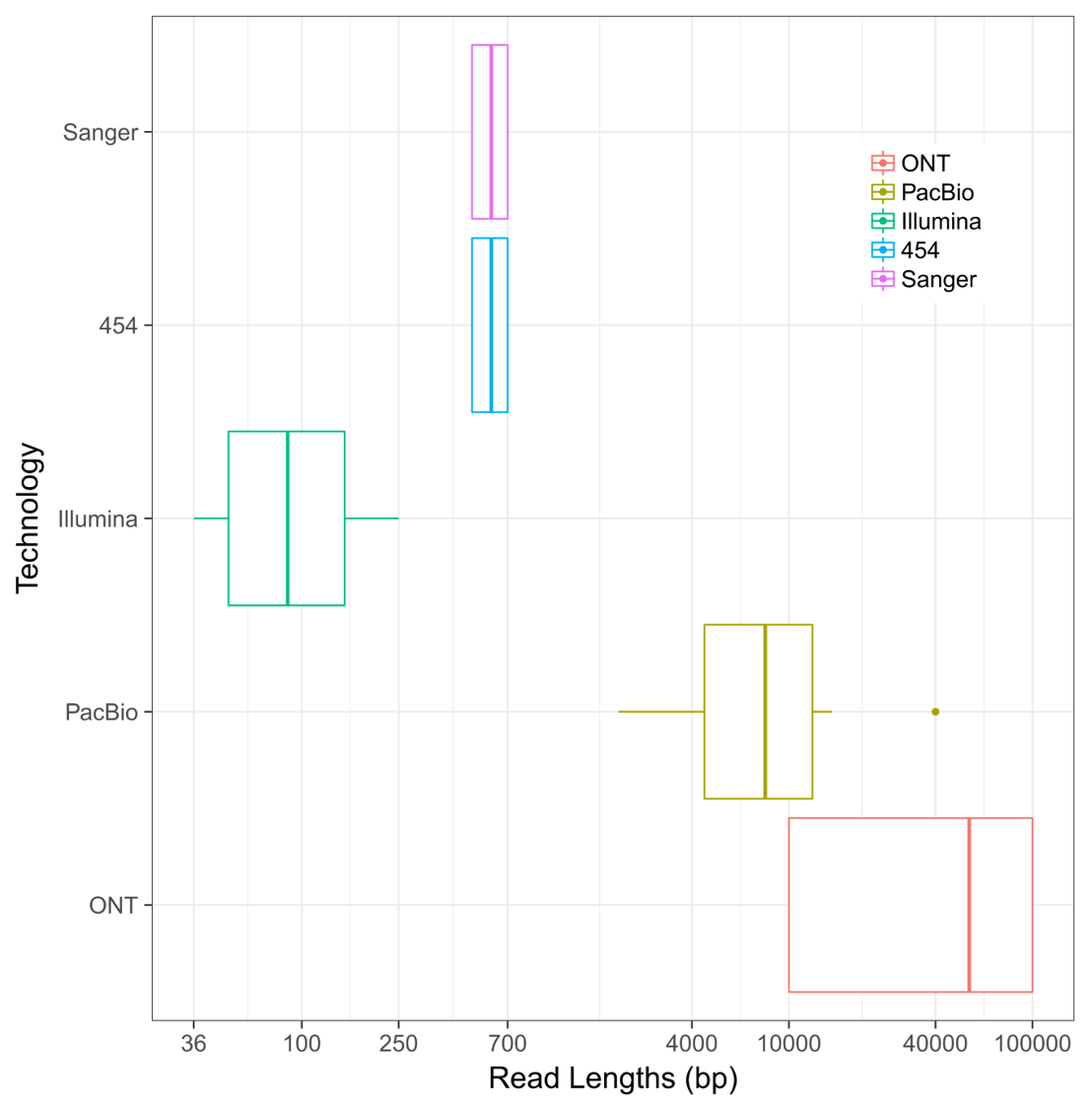
The research community knew that having longer sequence reads would solve one of the biggest problems in genome assembly: dealing with large (> 1000 bp) repeats. While they may not seem to be a major concern at first, repetitive DNA elements cause ambiguity in sequence read assembly. If a read from a repetitive region could occur from 5+ equally likely regions in the genome and there is no unique sequence to anchor it from both ends, the assembly of that region “stops.” This creates highly fragmented reference genomes by virtue of the fact that these elements could not be confidently spanned!